
El Síndrome de Angelman es un trastorno del neurodesarrollo de origen genético que se caracteriza por un retraso en el desarrollo de la persona. Este síndrome tiene lugar cuando se produce una carencia del funcionamiento de ciertos genes del cromosoma 15, sea por mutaciones o bien por deleciones por herencia materna.
Es un síndrome que no se empieza a hacer visible hasta que el bebé tiene, aproximadamente, entre 6 y 12 meses, momento en el que tendría que iniciar un desarrollo normal.
Actualmente, la edad de diagnóstico es cada vez más precoz y, en muchos casos, se da antes de los 2 años de edad gracias a la divulgación y conocimiento del síndrome por parte de los profesionales y a las avanzadas técnicas de análisis genéticos.
¿Cuáles son las causas?
Las principales causas del Síndrome de Angelman están relacionadas con la interrupción, inactivación o ausencia del gen UBE3A (ubicado en el cromosoma 15 materno).
El tipo de causa determinará el grado de afectación (más o menos severa). De esta forma:
Delección del cromosoma materno: esta es la causa más frecuente del Síndrome de Angelman (hasta el 70% de los casos). Consiste en una pérdida de un trozo de cromosoma, que se rompe y se separa del resto del material genético. En este caso, la afectación del niño o de la niña es bastante severa y hay presencia de convulsiones, relativa hipopigmentación, dificultades motoras, ataxia, hipotonía muscular, dificultades en la alimentación, retraso cognitivo y discapacidad en el habla.
Disomía uniparental (UPD): en un pequeño porcentaje de los casos, el SA se produce cuando se heredan dos cromosomas 15 del padre y ninguno de la madre. La afectación suele ser menos severa, ya que el desarrollo físico es mejor y no hay tanta afectación en la coordinación y en los movimientos, y la presencia de convulsiones es menor.
Defectos en el centro de la impronta o de la mutación en el gen UBE3A, que deriva del cromosoma 15 de la madre: en estos casos, la afectación clínica no suele ser tan severa y tienden a ser el grupo de afectados con unas mayores habilidades cognitivas, lenguaje receptivo y mayor habilidad en la motricidad fina y gruesa.

¿A quién afecta?
No se conoce con exactitud la incidencia de este síndrome entre la población mundial. Diferentes estudios la sitúan entre 1 de cada 12.000 y 1 de cada 24.000 recién nacidos y es por eso que se considera como “enfermedad rara”. Afecta por igual a ambos sexos y no existe predominio en una raza en concreto.
¿Cuáles son las características o síntomas?
- Retraso en el desarrollo funcionalmente severo.
- Retraso cognitivo.
- Problemas de movimiento y de equilibrio, principalmente ataxia al caminar (rigidez, torpeza y pérdida de coordinación) y movimientos temblorosos de las extremidades.
- Conducta muy característica en el comportamiento: cualquier combinación de risa o sonrisa frecuente, apariencia feliz, personalidad fácilmente excitable, a menudo con aleteo de manos, hiperactividad, permanencia de la atención durante poco tiempo.
- Problemas en el habla: no existe lenguaje oral o hay un uso mínimo de palabras. Las habilidades de comunicación receptivas (es decir, la comprensión) y no verbales son mayores que las verbales.
Otras características frecuentes son:
- Retraso o crecimiento inferior al esperado de la circunferencia de la cabeza (perímetro cefálico).
- Crisis convulsivas (epilepsia) que comienzan habitualmente antes de los 3 años de edad. La gravedad de las convulsiones disminuye con la edad, pero permanecen durante la vida adulta.
- Electroencefalograma anormal, con un patrón característico (con ondas de gran amplitud y picos lentos).
Además, pueden aparecer otras características asociadas como:
- Occipucio plano (esto es, la parte posterior-inferior de la cabeza achatada).
- Lengua prominente.
- Problemas para succionar y tragar.
- Problemas de alimentación durante la infancia.
- Hipotonía troncal durante la infancia.
- Prognatismo (esto es, mandíbula prominente).
- Boca ancha y con los dientes espaciados.
- Babeo frecuente.
- Conductas excesivas de mascar/masticar.
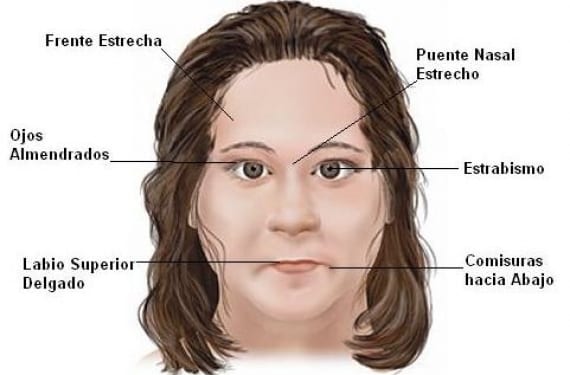
- Estrabismo.
- Piel hipopigmentada (clara), cabello claro y ojos también claros (en comparación con la familia).
- Reflejos hiperactivos de los tendones de las extremidades inferiores.
- Brazos levantados, flexionados, especialmente al caminar.
- Hipersensibilidad al calor.
- Trastornos del sueño: ciclos anormales de sueño, despertares frecuentes y una disminución de la necesidad de dormir.
- Atracción hasta la fascinación por el agua. También por objetos crujientes, tales como cierta clase de papeles o plásticos.
- Escoliosis.
- Estreñimiento.
¿Tiene cura este trastorno?
En la actualidad, el Síndrome de Angelman no tiene cura, aunque existen varias líneas de investigación al respecto. Estas investigaciones están reportando resultados muy esperanzadores para la comunidad Angelman.
Al ser una enfermedad minoritaria, la investigación es financiada en gran medida por iniciativas privadas de familias y empresas interesadas en encontrar la cura para los afectados, o algún tratamiento que mejore la calidad de vida de los pacientes y de sus familiares.
Fuente: Asociación Síndrome de Angelman (ASA)



